Few problems in the LC-MS lab are as demoralizing as watching your signal quietly disappear. A method that gave clean, intense peaks last month now barely clears the noise — same sample, same instrument, no obvious change. Because an LC-MS signal depends on the entire chain from sample prep through chromatography to ionization and detection, “lost sensitivity” can originate almost anywhere.
This guide gives you a structured way to localize signal loss instead of changing variables at random. The core question to answer first: is the analyte not getting to the source, not ionizing, or not being detected?
Step 1: Separate a gradual decline from a sudden drop
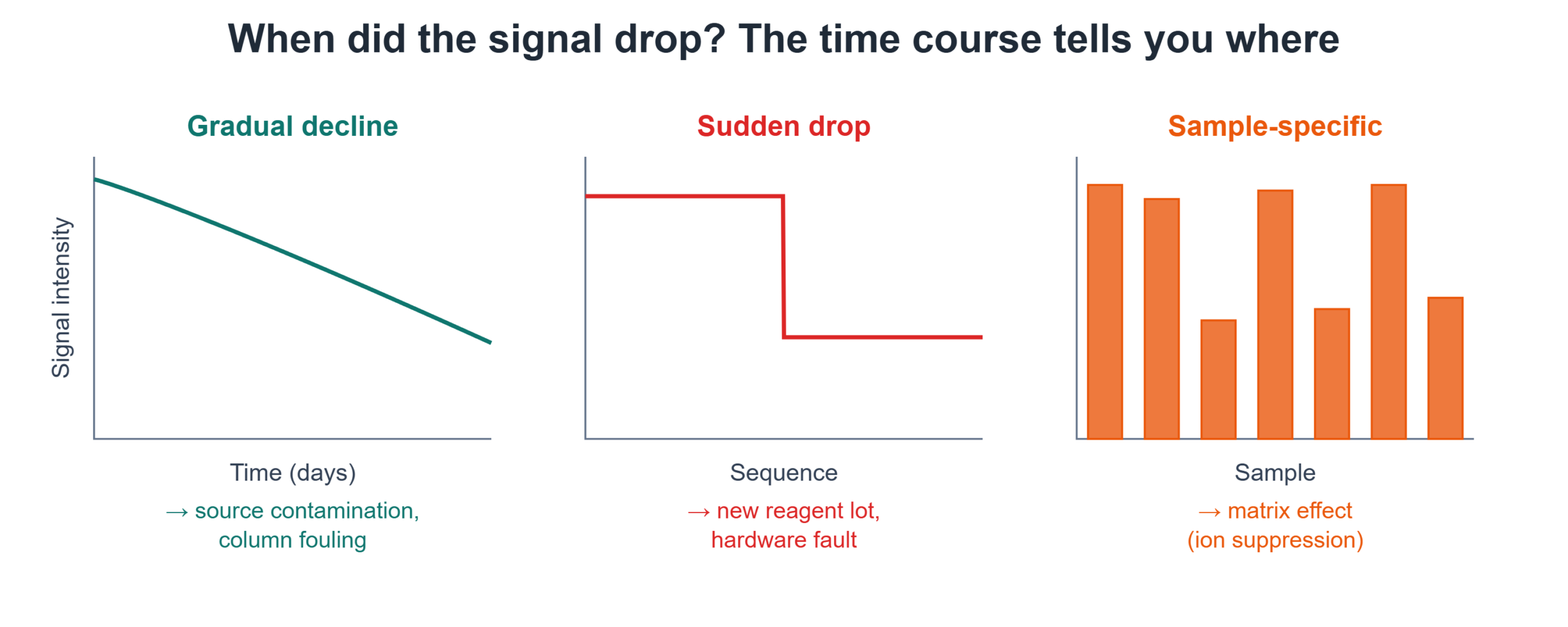
The time course of the problem is your most powerful early clue.
- Gradual decline over days or weeks points to accumulation: source contamination, capillary fouling, or slow column degradation.
- Sudden drop between one sequence and the next points to a discrete event: a new mobile phase batch, a fresh reagent lot, a changed sample matrix, or a hardware fault.
- Drop confined to certain samples points to the matrix — ion suppression from co-eluting interferences.
Pin down which of these you’re dealing with before touching the instrument.
Step 2: Ion suppression — the most common and most overlooked cause
Ion suppression — the matrix effect that most often undermines quantitative LC-MS and LC-MS/MS bioanalysis — is the silent killer of ESI sensitivity. Co-eluting matrix components — phospholipids, salts, detergents, and endogenous compounds — compete with your analyte for charge and droplet surface during electrospray, reducing the analyte’s ionization efficiency without ever appearing as a visible peak. The chromatogram can look clean while your signal is quietly halved.
How to confirm it:
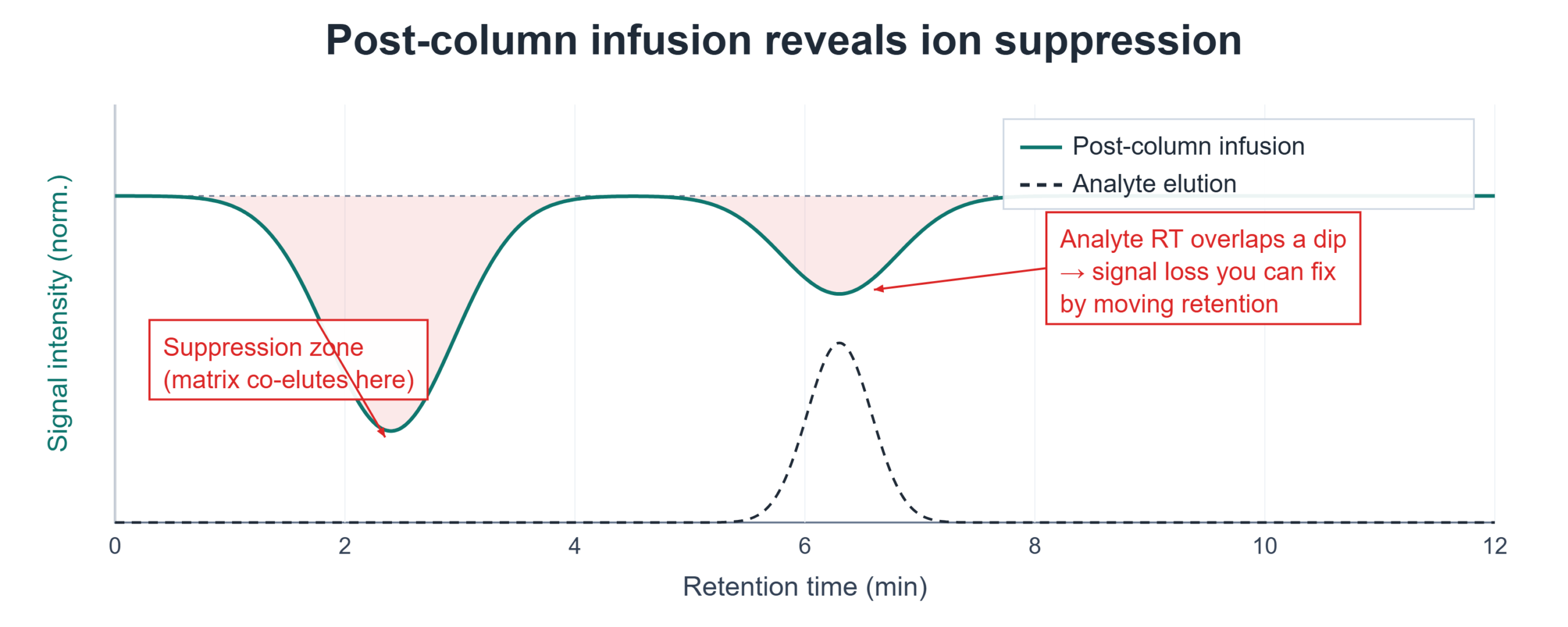
- Post-column infusion. Infuse a steady stream of your analyte through a tee post-column while injecting a blank extract of your matrix. Watch the infused signal: where it dips is where matrix components are suppressing ionization. If the dip overlaps your analyte’s retention time, you have a suppression problem to engineer around.
- Compare matrix vs. neat. Spike your analyte into extracted blank matrix and into neat solvent at the same concentration. A lower response in matrix quantifies the suppression directly.
The fixes are chromatographic and preparative: move the analyte’s retention time away from the suppression window, improve sample cleanup (protein precipitation → solid-phase or phospholipid-removal cleanup), or dilute the matrix load. Suppression is rarely fixed at the instrument; it’s fixed upstream.
Step 3: Source contamination and fouling
If the decline is gradual and affects everything, the source is a prime suspect. Deposits build up on the sampling cone/capillary, sprayer, and ion optics, progressively robbing transmission. The signature is a slow, across-the-board sensitivity loss that no method change reverses. Cleaning the source components on the manufacturer’s schedule — and more often with dirty matrices — usually restores it. Track your sensitivity over time so you can see the decline before it becomes a crisis.
Step 4: Mobile phase, reagents, and additives
A sudden drop that coincides with a fresh bottle is a strong signal. Common culprits: a new lot of mobile phase additive at the wrong concentration, a contaminated solvent bottle, degraded or wrong-pH buffer, or an additive that suppresses ionization (for example, too much of a non-volatile or ion-pairing agent). Always keep one bottle of known-good mobile phase to swap in as a control — it isolates a reagent problem in a single injection.
Step 5: Chromatographic changes masquerading as sensitivity loss
Sometimes the ionization is fine and the peak has changed. A broadened peak spreads the same amount of analyte over more time, lowering peak height even when the integrated area is unchanged. Check area versus height: if area is stable but height fell, you have a broadening/retention problem, not a true sensitivity loss — investigate the column and gradient instead of the source.
Step 6: Analyte and standard stability
Before blaming the instrument, rule out the sample. Degraded standards, adsorptive losses in vials or tips, or an unstable analyte in the prepared matrix all present as “lost signal.” Re-prepare from fresh stock and re-inject. If fresh standards restore the response, the instrument was never the problem.
A localization checklist
- Characterize the time course — gradual (source, column) vs. sudden (reagents, hardware) vs. sample-specific (matrix suppression).
- Check area vs. height — separates true signal loss from peak broadening.
- Run post-column infusion if the loss tracks with certain matrices — confirms suppression.
- Swap in known-good mobile phase — isolates reagent problems fast.
- Re-prepare standards from fresh stock — rules out sample stability.
- Clean the source if the decline is gradual and universal.
The takeaway
Lost LC-MS sensitivity feels like a single problem but is really a family of problems separated by where in the chain the signal is being lost. The fastest path to the answer is to characterize when and for which samples the signal dropped, then use targeted tests — post-column infusion for suppression, area-versus-height for broadening, fresh standards for stability — to confirm the mechanism before you start cleaning hardware or rebuilding methods.
Related troubleshooting guides: Autosampler carryover in HPLC and LC-MS and Ghost peaks in HPLC.
For a guided diagnostic on peak integrity and signal loss in quantitative LC–MS, try the LabVeda LC–MS PK Peak Integrity Engine.